


新药研发
使命赋创新以动力,创新赋生命以可能
医药资讯
【深度报告】BCR-ABL1变构抑制剂,开启靶向治疗新篇章
2021-06-16 浏览量:
变构调节(allosteric regulation)在细胞生命过程中普遍存在,包括分子间非共价相互作用、共价修饰(磷酸化、点突变,以及与小分子的化学反应)、细胞环境(温度、辐射、pH和离子强度)和光吸收等[1–4]。变构效应精确控制着细胞的正常生命过程,包括细胞信号转导、代谢、酶的催化以及基因调控。蛋白变构调节失调与许多疾病密切相关,如癌症、精神障碍、糖尿病、炎症和免疫疾病等[5-7]。
变构药物是指通过与靶蛋白上的变构位点(空间上不同于常规活性位点或底物结合位点等正构位点)相结合,从而调控蛋白生物功能的效应分子[5,8,9]。这类新型调节剂可能通过两种方式影响蛋白的生物效应。一种是变构调节引发变构位点及其邻近原子发生扰动并重新定向,从而产生应变能并迫使下一层原子移动,进而影响下一层原子。变构扰动通过变构波形式从变构位点传播到正构位点,导致正构位点构象动力学微调[1,6,10]。另一种是最近提出的动力学驱动的变构概念,变构扰动遍及整个蛋白结构(包括正构位点在内),改变整个蛋白的振动模式和构象群体,可引发蛋白构象发生重大变化[11,12]。
一、变构调节剂是研发新风向
相对于传统的正构(orthosteric)药物,变构药物具有几个显著优势:1)克服耐药性:在抗菌、抗病毒和抗肿瘤治疗中,耐药性问题一直是个难题,变构调节剂与正构调节剂作用于靶点的不同位点,基于不同的作用机制发挥药效,这使变构调节剂有可能克服正构调节剂在治疗过程中产生的获得性耐药[13];2)高选择性:变构位点的同源序列较低,氨基酸残基保守性低,因此具有更高的选择性、更低的脱靶毒性和更低的给药剂量[14];3)解决难成药靶点困境:有些正构位点口袋残基高度保守,且为极性和带电荷环境,不仅导致调节剂缺乏选择性、毒性高,而且导致调节剂的细胞渗透性和口服生物利用度差而缺乏成药性;其次,正构位点的界面大且平坦,缺少合适的小分子结合口袋(如蛋白相互作用界面类靶点);又或者天然底物(ATP/GTP)在细胞中的浓度非常高,且与活性位点的亲和力非常强,使得直接靶向正构位点的调节剂极难发挥作用等[15-16];4)变构调控可能更精准:有些蛋白功能窗口较小,既需要调控又不能过度调控,而变构调节剂可以双向调节蛋白功能,发挥精准调控作用,如变构激动剂更不容易诱导蛋白受到激活作用后脱敏[17]。
由于变构调节药物显示出巨大的开发前景及其较传统正构药物的显著优势,靶点变构效应已被确立为药物发现的新机制,引发了变构药物发现、优化以及临床开发的热潮,吸引了众多生物科技公司和大型制药企业涌入这一热门领域,触发了许多大额融资、合作与交易[18]。以下,我们将以BCR-ABL1变构抑制剂为例,全面介绍这类新型药物的发展历史、作用机制、临床进展、应用潜力等多方面内容。
二、BCR-ABL1抑制剂发展历史
伊马替尼作为首个上市的靶向治疗药物,开创了小分子靶向治疗肿瘤的时代,被美国《时代》周刊誉为”银色子弹“。这款BCR-ABL1抑制剂的成功研发使得原来致死率很高的慢性粒细胞白血病(CML)变成了慢性病,CML的治疗也因此获得了里程碑式的进步[19]。然而,10%~20%的CML患者在接受伊马替尼治疗后产生了获得性耐药,主要原因是BCR-ABL1基因可产生50多种点突变[20]。虽然后续几种二代药物的上市使CML的治疗方案得以革新,为伊马替尼耐药的CML患者带来了更多的临床获益,但是大多数患者仍须持续用药,而且最棘手且最常见的耐药突变BCR-ABL1T315I对目前所有一、二代BCR-ABL1抑制剂均耐药。尽管在国外上市的泊那替尼(Ponatinib)可以克服BCR-ABL1T315I耐药,但该药会引发与治疗相关的动脉血栓形成和肝脏毒性风险,并且体外实验显示,一些少见的单点突变以及多重突变(compound mutations)也会对泊那替尼产生耐药[21]。目前,仍有多家国内外药企在积极开发第三代BCR-ABL1抑制剂,但它们依然是通过与ATP竞争活性位点的正构抑制剂,且结构与泊那替尼相似,因此与泊那替尼类似的安全性以及交叉耐药(cross resistance)问题依然存在。
为克服上述BCR-ABL1T315I以及对泊那替尼单点或多重突变耐药、解决现有药物的不耐受性和毒性问题,制药巨头诺华率先布局了第四代BCR-ABL1变构抑制剂(ABL001),目前该项目正在开展III期临床研究;国内方面,进展最快的是深圳市塔吉瑞生物医药有限公司(以下简称“塔吉瑞”)的TGRX-678,该BCR-ABL1变构抑制剂已经进入临床开发阶段。
三、BCR-ABL1变构抑制剂的作用机制
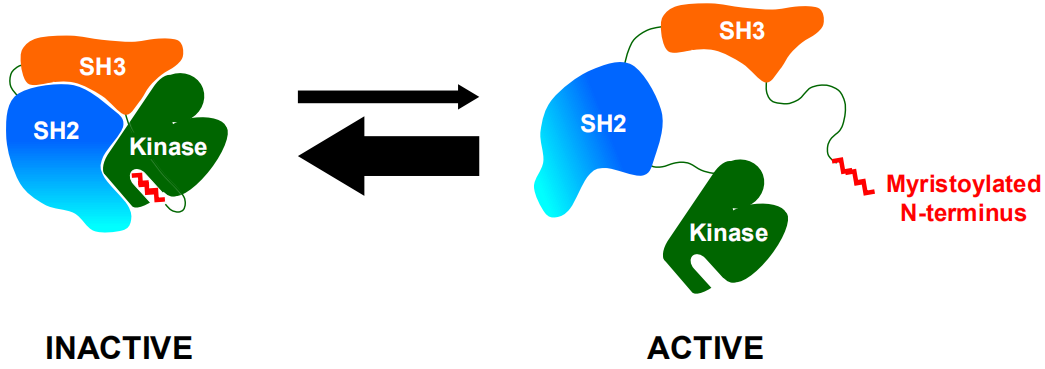
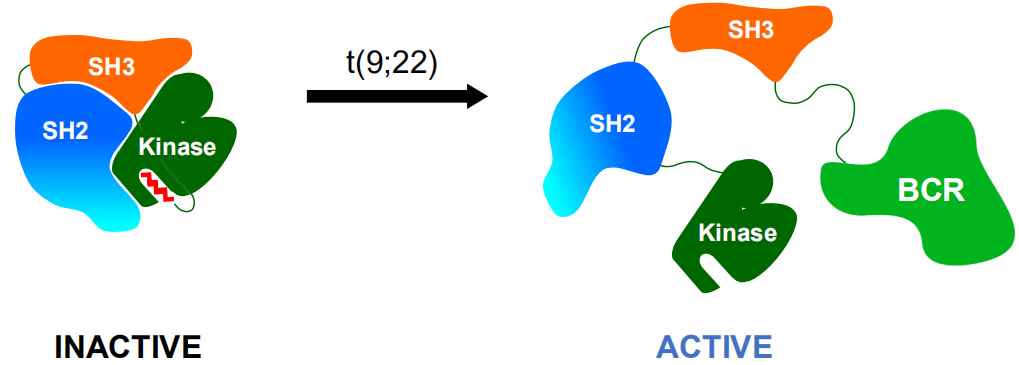
通常情况下,ABL1激酶域的变构位点被其自身N端肉豆蔻酰肽占据,诱导SH3-SH2-Kinase结构域发生交联,可发挥负向调控ABL1激酶活性的关键作用,调控着细胞正常增殖信号的传导(图1)。当BCR与ABL1基因融合后,BCR-ABL1激酶的N端肉豆蔻酰肽丢失,导致ABL1激酶域上的变构位点空缺,无法诱导SH3-SH2-Kinase结构域交联,自抑制平衡被打破,处于活化开放构象,致使BCR-ABL1激酶持续激活,诱发细胞增殖和肿瘤形成(图2)[22,23]。
为了恢复ABL1激酶负向调控功能,诺华和塔吉瑞通过模拟天然ABL1激酶肉豆蔻酰N端肽的功能,分别开发了ABL001 和TGRX-678。这两个分子与激酶结构域C端肉豆蔻酰空缺变构位点结合(图3a紫色球形区域),使得SH3和SH2结构域交联到激酶结构域,将ABL1激酶由活性致癌状态转变为“封闭”失活非致癌状态,重新稳定了ABL1激酶的自抑制构象(图3),从而发挥抗肿瘤作用[24]。

由此可见,第四代BCR-ABL1抑制剂的作用位点和机制与前三代正构抑制剂截然不同。第一代抑制剂伊马替尼、第二代抑制剂达沙替尼、尼洛替尼、博苏替尼、拉多替尼、氟马替尼,以及第三代抑制剂泊那替尼的分子机制是与ATP结合位点“铰链区”结合(图3a红色球形区域),为ATP竞争性抑制剂,抑制BCR-ABL1激酶的自身磷酸化和底物磷酸化,从而抑制癌细胞的增殖和肿瘤形成,均为典型的正构抑制剂。ABL001和TGRX-678 作用于BCR-ABL1激酶域的变构位点,其位于ABL1激酶催化结构域的C端肉豆蔻酰结合位点(图3a紫色球形区域),远离正构ATP活性位点及T315I突变氨基酸残基,因此ABL001和TGRX-678 与BCR-ABL1结合几乎不受T315I的影响。
ABL001和TGRX-678 的变构调节作用不仅能够通过变构“波”的形式引发正构位点构象动力学微调,使得对T315I耐药无效的尼洛替尼重新与BCR-ABL1T315I激酶正构位点结合,恢复其抑制作用。此外,变构抑制剂动力学驱动的变构效应,导致SH3和SH2结构域交联到激酶结构域,使整个蛋白发生重大构象变化,从而形成失活非致癌状态。尽管BCR-ABL1Y253H/T315I多重突变对泊那替尼耐药,但有研究提出泊那替尼与ABL001联用时,泊那替尼可短暂性占据正构活性位点,诱导构象从DFG-in活性构象转变为DFG-out非活性构象,这种构象的改变大大降低了肉豆蔻酰变构位点的柔性,使其能够组装并与ABL001更好的结合。反过来,ABL001与变构位点结合后预计将导致泊那替尼与BCR-ABL1激酶的DFG-out非活性构象进一步持续、稳定的结合,这种”正构-变构“相互协同的调节作用机制,最终导致强有力的激酶抑制,以克服对泊那替尼的突变耐药性[21]。因此,正构抑制剂与变构抑制剂联合可发挥强大的协同作用,产生意想不到的疗效。
四、BCR-ABL抑制剂临床前及临床试验结果
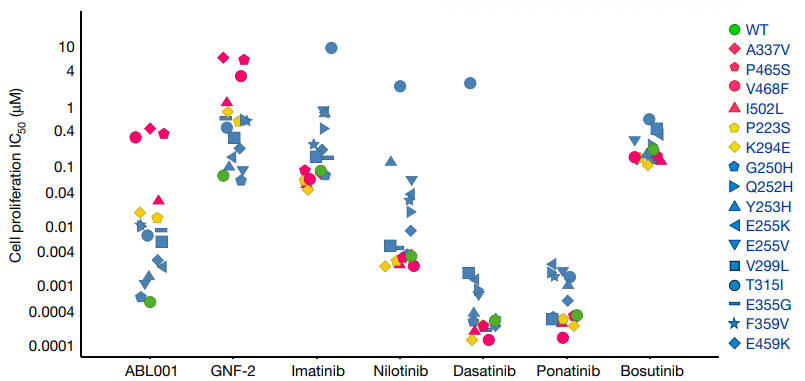
临床前研究表明,0.1nM级别以上的ABL001无论在ATP低浓度(10uM)还是高浓度(2mM) 条件下,对激酶的抑制率相当,表明ABL001是有效的BCR-ABL1非ATP竞争性激酶抑制剂。ABL001对BCR-ABL1野生型、T315I及其它突变型的Ba/F3细胞都有较强的生物活性(图4),可有效针对正构抑制剂引起的获得耐药性问题,特别是第一、二代无法克服的守门残基T315I突变耐药[24]。
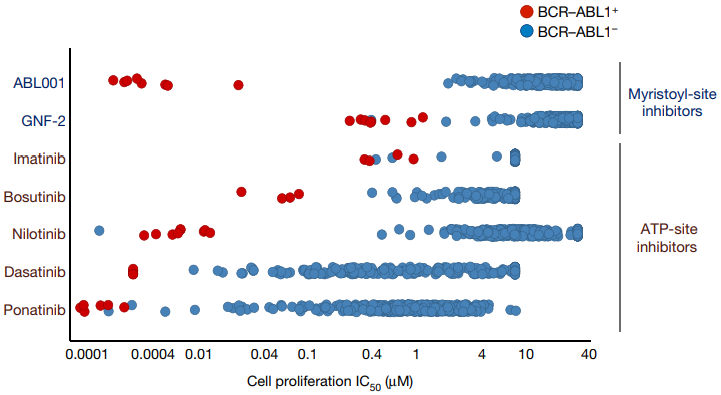
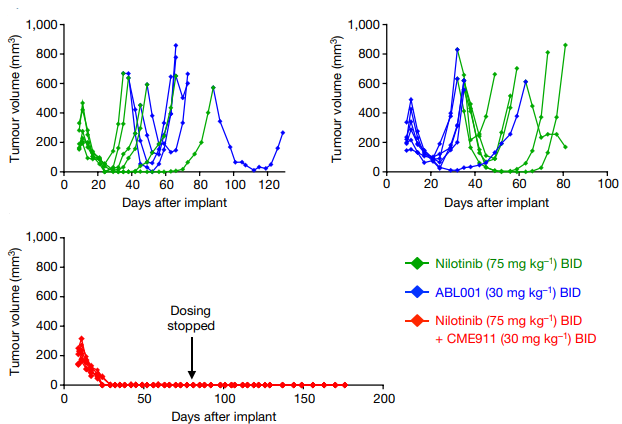
此外,多种实验对ABL001的选择性进行了评估。在ABL001浓度<10uM下,不会抑制其它60多种重组激酶的磷酸化作用[24]。采用450种肿瘤细胞,对ABL001变构抑制剂和正构抑制的活性和选择性进行评估发现,ABL001对BCR-ABL1阳性肿瘤细胞表现出优异的抗增殖活性(IC50=1-20nM),而对BCR-ABL1阴性肿瘤细胞的抑制活性较低(IC50>1000nM),并且其选择性优于正构抑制剂,如图5所示。此外,浓度<3uM的ABL001对G蛋白偶联受体、细胞转运蛋白、离子通道、核受体和酶无实质性影响[25]。ABL001不仅克服第一、二代抑制剂的耐药性,还凸显出其高度的选择性,可能避免出现第三代抑制剂泊那替尼引起的严重毒副作用。临床前研究还表明,ABL001与尼洛替尼联用,这一互补组合达到了很强的治疗效果,可完全控制异种移植肿瘤小鼠的肿瘤进展,且停止给药后不易复发(图6)[24]。ABL001与泊那替尼联用,不仅对泊那替尼最强耐药的多重突变Y253H/T315I、E255V/T315I等产生活性,还能有效抑制BCR-ABL1多重突变的出现并逆转泊那替尼的耐药,系统地解决了因多重突变造成的耐药性问题,且有望大大降低临床所需泊那替尼的浓度,减少副作用,为CML患者提供一种有效的治疗策略[21]。
临床研究表明,在对既往正构抑制剂耐药或发生不可接受的副作用,并且接受过至少两种正构激酶抑制剂治疗的CML患者,包括泊那替尼治疗失败的患者和有T315I突变的患者中,ABL001表现出较好的安全性和有效性[26]。ABL001与伊马替尼联合应用的临床试验表现出有希望的初步疗效,且具有良好的安全性和耐受性[27]。ABL001的一项III期临床试验ASCEMBL结果显示其与博苏替尼(bosutinib)相比,24周的主要分子学缓解(MMR)率显著且更优,研究达到主要终点[28]。研究人员正在评估ABL001与其它正构激酶抑制剂联合用作一线CML治疗方案。ABL001在治疗CML的临床试验中已初显成效,预示着显著的临床药用价值。
TGRX-678是国内首个且研发进展最快的第四代BCR-ABL1变构抑制剂,也是全球第二款BCR-ABL1变构抑制剂,目前已获得中国临床许可(受理号:CXHL2000158/CXHL2000159),正在积极筹备临床试验。临床前体内外研究结果显示,与ABL001相比,TGRX-678对Ba/F3 Bcr-AblT315I细胞的活性和选择性更高,口服生物利用度更佳,动物体内安全性也优于ABL001,有潜力成为best-in-class,因此其在临床上的表现令人期待。
五、总结与展望
基于独特的作用位点和机制,BCR-ABL1变构抑制剂不仅可以用于治疗既往对正构抑制剂耐药(包括守门残基T315I突变耐药)或不耐受的CML患者,与正构抑制剂的联合应用还能够克服泊那替尼单点和多重耐药,降低耐药性的发生,逆转药物的获得耐药性,减少给药剂量,降低毒副作用,有望使患者长期维持无治疗缓解,实现功能性治愈。BCR-ABL1变构抑制剂还有望用于治疗其它肿瘤或疾病,大幅拓展其临床适应症和药用价值。期待这类新型药物能够开启靶向治疗的新篇章,为更多克服耐药性药物的研发指引新方向。
参考文献:
上一篇:塔吉瑞全新药物设计及高端研发平台
下一篇:最后一页

 联系电话:+86-0755-86934300
联系电话:+86-0755-86934300 地址:深圳市南山区科兴科学园 A1 单元 3 楼
地址:深圳市南山区科兴科学园 A1 单元 3 楼 邮箱:
邮箱: